
Ciliary membrane complex for photoreceptor protein transport
Resource: State Key Laboratory of Ophthalmology
Proofread by: Jiawei Wang
Edited by: Xianjing Wei
Reviewed by: Xiaoling Liang
Visual information is essential for humans and animals to survive, and 80-90% of external information comes from vision. The acquisition of visual information begins with the capture of photons by photoreceptors. The membrane discs of the photoreceptor outer segment (OS) capture photons through opsin proteins, which initiate the phototransduction cascades. The membrane discs, renewed about 10% every day, require large amounts of lipids and proteins. However, the OS does not have the anabolic machinery, and the components for phototransduction must be continuously transported through a narrow bridge (connecting cilium (CC)) that connects to the inner segment (IS) where biomolecules are synthesized. Failure in protein transport to the OS can lead to protein accumulation within the IS, causing stress-induced photoreceptor death and triggering degenerative retinal blinding diseases, for instance, the Retinitis pigmentosa (RP). The estimated incidence of hereditary RP is about 1:4000~5000 worldwide, affecting nearly 300,000 people in China. And many genes mutated in RP are related to OS protein transport of the photoreceptor cilia.
Despite many functional studies on protein transport of the photoreceptor cilia, most of these have focused on the microtubules and associated structural proteins, motors (dyneins), and the intraciliary vesicular trafficking machinery (IFTs), but the role of ciliary membrane proteins of the CC is unclear.
On April 10, 2022, Chunqiao Liu and Yizhi Liu's groups from Zhongshan Ophthalmic Center of Sun Yat-sen University jointly published a research paper, "Tmem138 is localized to the connecting cilium essential for rhodopsin localization and outer segment biogenesis" in Proceedings of the National Academy of Sciences (PNAS). This study reveals for the first time that the photoreceptor ciliary membrane complex plays an essential role in the trafficking of Rhodopsin and other OS proteins, thus providing a new line of mechanisms complementary to that existing for RP diseases. The study additionally provides valuable information for the molecular mechanisms of hydrocephalus of the central nervous system caused by ciliopathy.
Liu's group has been focusing on developmental and genetic diseases of the neural retina. Previous studies on the Wnt/PCP pathway involved in ciliary development and cell polarity found several regulated transmembrane proteins. Among them, mutation of human TMEM138 causes Joubert syndrome with retinal dystrophy. To understand the pathogenetic mechanism of retinal phenotype caused by TMEM138 mutation, the authors first created a Tmem138 knockout mouse model, which successfully recapitulated the phenotype of Joubert syndrome, showing enlarged brain ventricles, azoospermia, and retinal degeneration. Germline deletion of Tmem138 resulted in failed photoreceptor OS outgrowth observed by immunofluorescence, transmission, and scanning electron microscopy (Fig. 1A-C).
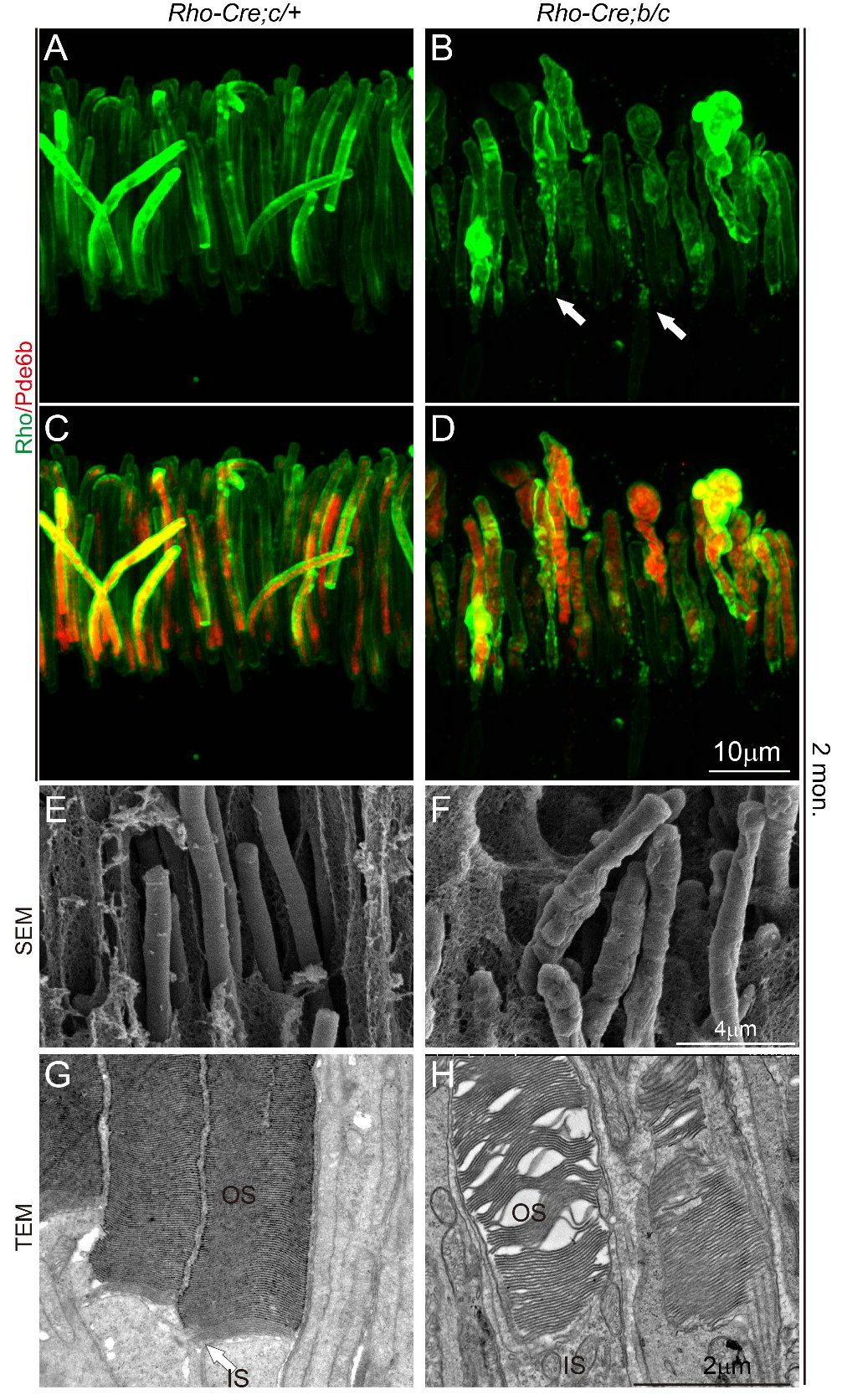
Fig. 1 Disruption of Tmem138 caused retinal degeneration
Failure to develop OS may be due to defects in any or all processes of cilia biogenesis, protein trafficking, or assembly of the OS membrane discs. The authors first examined the formation of OS in mutant mice by electron microscopy and confirmed that the cilia structure was normal. Furthermore, immunofluorescent labeling showed that the localization of IFT subunits and dynein (IFT88, Kif3A)-dependent microtubule transporting components did not alter significantly, and membrane disc assembly proteins (Rds/Prominin-1/Prcad) were also relatively normal in the nascent OS. These experiments generally rule out the possibility of defective initiation of developmental cilia and outer segment membrane disc.
Next, the authors utilized multiple immunofluorescent labeling to define different domains of OS cilia in both normal and knockout photoreceptors and located Tmem138 to the proximal CC (Fig. 2A, B). The loss of the distal Ahi1 domain of the mutant CC suggests that Tmem138 is closely related to the intracellularly localized Ahi1. Consistent with this observation, mutations of human AHI1 also cause Joubert syndrome and fail to develop photoreceptor OS. These results suggest that membrane protein Tmem138 is likely involved in the trafficking of OS proteins together with Ahi1. After examining a range of phototransduction components and OS structural proteins, Rhodopsin turned out to be the first aberrantly localized, coinciding with the onset of OS development.
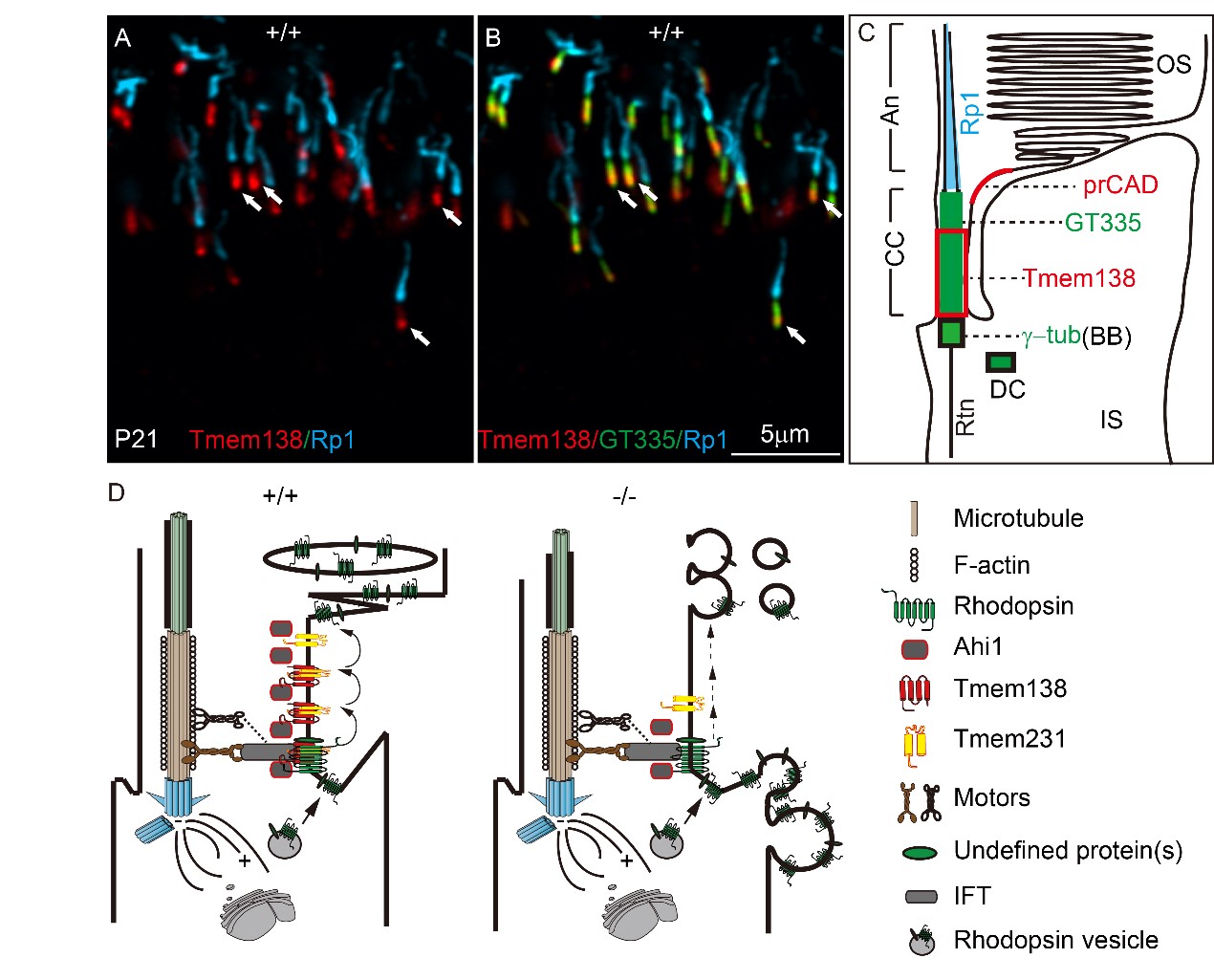
Fig. 2 Tmem138 localization of and its CC membrane complex
Following the observed mislocalization of Rhodopsin and Ahi1, the authors found that Tmem138 directly interacted with Ahi1 and Rhodopsin by co-immunoprecipitation and immuno-pulldown assays using either cultured cell or OS protein extracts. Furthermore, another transmembrane protein, Tmem231, interacts with Tmem138 and Rhodopsin reciprocally, and the ciliary membrane localization of Tmem231 was also altered in Tmem138 mutant mice.
Altogether, the authors propose that the Tmem138, Tmem231, and Ahi1 form a CC membrane complex playing a crucial role in the transciliary trafficking of Rhodopsin and other outer segment proteins (Fig. 2C). This is the first study to date describing the photoreceptor OS transport involving residential CC membrane proteins/complex.
The study was supported by the National Natural Science Foundation of China, the Guangzhou Science and Technology Innovation Project, and the Sun Yat-sen University Hundred Talents Program. Postdoctoral fellow Dr. Guo Dianlei, and Dr. Ru Jiali are the first authors. Professor Yanhong Wei from the School of Public Health of Sun Yat-sen University and Professor Xialin Liu from Zhongshan Ophthalmic Center contributed to the research. Professors Chunqiao Liu and Yizhi Liu are the corresponding authors of the paper.
Link to the article: https://www.pnas.org/doi/10.1073/pnas.2109934119